MIT
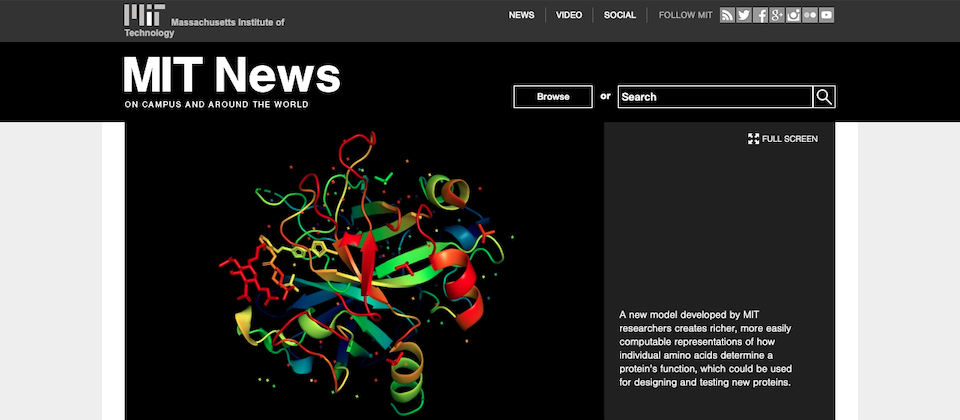
來自麻省理工學院的研究團隊近來透過機器學習技術,打造利用胺基酸鏈段預測蛋白質結構的模型,也就是能夠理解個別胺基酸鏈段如何決定蛋白質功能,對於生物相關研究、藥物開發、蛋白質設計和測試都是一大進展,未來,該機器學習模型能夠讓研究人員更專注於修改特定胺基酸片段,改善蛋白質工程。
蛋白質是由胺基酸分子線性鏈結而成,個別胺基酸分子透過肽鍵連接,根據鏈結中的物理相互作用和序列,折疊成相當複雜的3D結構,而這些不同的結構決定了蛋白質在生物學上的功能,因此,了解蛋白質的3D結構對於預測蛋白質對特定藥物的反應是有價值的。
過去數十年來的研究,主要都是用多種成像技術來研究蛋白質結構,而這些研究只能了解非常小部分的蛋白質結構,近來,研究人員開始用機器學習模型根據胺基酸序列,來預測蛋白質結構,但是,由於胺基酸序列的多元性,會生成非常類似的結構,再者,也沒有足夠多的結構樣本來訓練模型。
因此,有別於過去直接預測結構的方法,MIT研究團隊將預測蛋白結構的資料轉譯成以數值表示的向量值,為此,團隊用已知的蛋白質結構近似度,來監督機器學習模型,讓模型學習特定氨基與蛋白質功能的關係,首先,利用3D蛋白質結構當作訓練指引,來簡單地計算每個胺基酸在蛋白質序列的位置,接著,再透過計算出的位置代表值當作機器學習模型輸入資料,根據每個胺基酸鏈段來預測蛋白質功能。
訓練過程總共使用了22,000個來自蛋白質結構資料庫SCOP的蛋白質資料,該資料庫包含數千個依照相似結構和胺基酸序列分類的蛋白質,團隊利用SCOP資料庫分類的類別,模型針對每對蛋白質,計算蛋白質結構真實相似分數,因此,每個蛋白質結構的向量會包含與其他序列相似程度的資訊,再根據該相似分數預測胺基酸序列的3D結構。最後模型會將比對預測的相似分數和SCOP真實的相似分數的結果,當作回傳到編譯器的訊號。
同時,模型還會針對每個胺基酸序列向量預測聯繫地圖(contact map),也就是呈現出每個胺基酸在蛋白質預測結構中,與其他胺基酸之間的距離,該模型還會將預測的聯繫地圖與SCOP資料庫的聯繫地圖拿來做比較,回饋到模型中,如此一來,能夠幫助模型學習胺基酸在蛋白質結構中正確的位置,進一步更新胺基酸功能。
熱門新聞

2026-02-11
</a> on <a href=\"https://unsplash.com/photos/blue-and-black-digital-wallpaper-uDO6NuH7WFU?utm_content=creditCopyText&utm_medium=referral&utm_source=unsplash\">Unsplash</a>")
2026-02-11
2026-02-09
2026-02-10
2026-02-10


2026-02-06

2026-02-10